
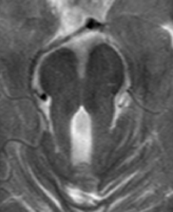
Das Joubert-Syndrom ist eine autosomal rezessiv vererbte Entwicklungsstörung, bestehend aus einer Kleinhirnwurm-Aplasie, einer cerebellären Ataxie, einer muskulären Hypotonie, einer okulomotorischen Apraxie, neonataler Tachypnoe, einer mentalen Retardierung und einer retinalen Degeneration. Es wird deshalb auch als cerebello-okulo-renales Syndrom bezeichnet. Neben den genannten Symptomen können eine Leberfibrose, okuläre Kolobome und eine Polydaktylie auftreten. Typischerweise stellt sich in der Magnetresonanztomographie (MRT) als Folge der Kleinhirnwurm-Aplasie das sogenannte „Molar Tooth Sign“ dar (Abb. 1). Eine Nephronophthise findet sich bei etwa 25% der Patienten mit Joubert-Syndrom. Dem Joubert-Syndrom liegen Mutationen im NPHP6/CEP290-Gen, das für Nephrozystin-6 kodiert oder im NPHP8/RPGRIP1L-Gen, das für Nephrozystin-8 kodiert, zugrunde. Weitere Mutationen konnten in anderen Genen identifiziert werden, darunter AHI1, MKS3, ARL13B, CC2DA2, INPP5E und TMEM216 [Übersicht in Wolf M, Pediatr Nephrol 2010]. Darüber hinaus wurden in wenigen Fällen bei Patienten mit Joubert-Syndrom auch Mutationen in NPHP1 und NPHP4 beschrieben.