
News
Das "Paper of the Month" 12/2024 geht an Niklas Hörsting, Kai Wohlgemuth und Heymut Omran von der Klinik für Kinder- und Jugendmedizin - Allgemeine Pädiatrie -
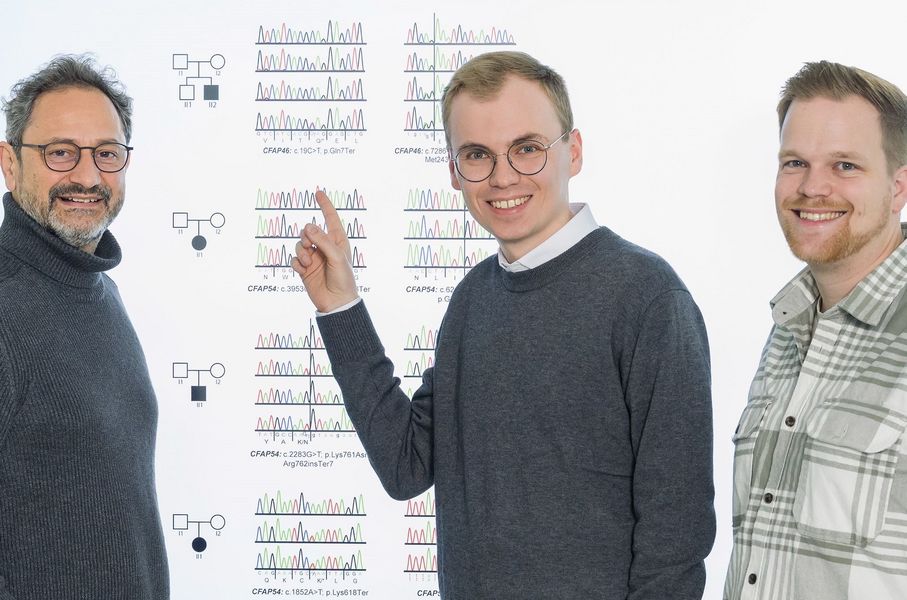
Prof. Heymut Omran, Niklas Hörsting, Kai Wohlgemuth (v.l.n.r.) sind die Autoren des neuen Papers of the Month (Foto: Uni MS/E. Wibberg)
Paper of the Month“ der Medizinischen Fakultät der Universität Münster an:
Niklas Hörsting, Kai Wohlgemuth und Heymut Omran von der Klinik für Kinder- und Jugendmedizin - Allgemeine Pädiatrie -
Wohlgemuth K; Hörsting N; (…); Omran H. Pathogenic variants in CFAP46, CFAP54, CFAP74 and CFAP221 cause primary ciliary dyskinesia with a defective C1d projection of the central apparatus. Eur Respir J. 2024 Dec 12;64(6):2400790.
Begründung der Auswahl:
Die primäre ziliäre Dyskinesie (PCD) ist eine seltene genetische Störung, die durch eine unzureichende mukoziliäre Clearance verursacht wird. In der vorliegenden Arbeit werden bislang nicht beschriebene pathogene Varianten in PCD-Genen beschrieben, die eine frühere genetische Untersuchung von PCD-Patienten nahelegen, als sie derzeit in Leitlinien empfohlen wird.
Zu Hintergrund, Fragestellung und Bedeutung der Publikation:
Die PCD äußert sich unter anderem in chronischen Atemwegsinfektionen. Die Leitlinie der Europäische Atemwegsgesellschaft empfiehlt die Bewertung der klinischen Historie, die Messung der nasalen NO-Produktionsrate, die Videomikroskopie-Analyse des ziliären Schlags sowie die TEM-Analyse der ziliären Ultrastruktur. Genetische Untersuchungen können als letzter Schritt durchgeführt werden.
Das Forschungsteam konnte erstmals zeigen, dass pathogene Varianten in den Genen CFAP46, CFAP54, CFAP74 und CFAP221 zu einer defekten C1d-Projektion des ziliären Zentralapparats, einem Bestandteil des respiratorischen Flimmerepithels, führen. Diese Gene können dadurch erstmals der PCD mit einer defekten C1d-Projektion zugeordnet werden. Außerdem lässt sich dieser PCD-Typ nicht anhand der aktuellen Leitlinie diagnostizieren: Trotz schwerer Symptomatik wird er vom PCD-Vorhersagescore PICADAR nicht erkannt. Zudem ist er mit normalen nasalen Stickstoffmonoxid-Produktionsraten, einer normalen ziliären Ultrastruktur und einem normalen Schlagmuster assoziiert. Doch durch neuartige in-vitro-Analysen der ziliären Transportkapazität konnte die insuffiziente Atemwegsreinigung dargestellt werden.
Die Studie erweitert einerseits das Spektrum bekannter PCD-Gene, andererseits betont sie, dass die PCD mit defekter C1d-Projektion dem aktuellen Diagnostik-Algorithmus entgeht. Die von der Arbeitsgruppe vorgeschlagene Priorisierung genetischer Untersuchungen wird für die Entwicklung einer neuen Leitlinie von enormer Relevanz sein.
Background and fundamental question of the publication:
PCD is a rare genetic disorder caused by insufficient mucociliary clearance leading to chronic airway infections. The diagnostic guideline of the European Respiratory Society recommends the evaluation of the clinical history, nasal nitric oxide (nNO) production rate measurements, high-speed video microscopy analysis of ciliary beating, and the assessment of ciliary axonemes via TEM. Genetic testing can be implemented as a last step.
The research team showed for the first time that pathogenic variants in CFAP46, CFAP54, CFAP74, and CFAP221 lead to PCD with a defective C1d projection of the central apparatus which is part of the respiratory epithelium. Careful evaluation revealed that C1d-defective PCD is associated with normal situs composition, normal nNO-production rates, normal ciliary ultrastructure by TEM, and normal ciliary beating by HSVMA. Despite chronic respiratory disease, PICADAR does not reliably detect this PCD type. However, it could be shown by in-vitro ciliary transport assays that affected individuals exhibit insufficient ciliary clearance.
Overall, this study extends the spectrum of PCD genes and highlights that C1d-defective PCD individuals elude diagnosis in the current diagnostic algorithm. To enable diagnosis, genetic testing should be prioritized in future diagnostic guidelines.
Förderung:
Die vorliegende Publikation wurde unter anderem durch das Förderinstrument Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Münster der Medizinischen Fakultät Münster (Projekte Om2/009/12, Om2/015/16, Om2/010/20; Projektleiter: Univ.-Prof. Dr. Heymut Omran) finanziell gefördert.
Die bisherigen ausgezeichneten „Papers of the Month“ finden Sie HIER.
Folgendes könnte Sie auch interessieren:

Münster (jr) – 10.000 Liter – das sind über 60 volle Badewannen und zugleich die beeindruckende Menge Luft, die unsere Lunge jeden Tag von Schmutz und Pollen reinigen muss. Liegt ein Defekt…

Münster (mfm/kd) – Der bereits mehrfach für seine Studien zu seltenen Krankheiten ausgezeichnete Medizinprofessor Heymut Omran von der Universität Münster kann sich über eine weitere Ehrung…

Münster (mfm/ab) - Der erfolgreiche Online-Start des ersten internationalen Patientenregisters für die seltene Lungenerkrankung PCD durch ein internationales Wissenschaftlerteam um Prof.…

Münster - Preisträger des diesjährigen Eva-Luise-Köhler-Forschungspreises für Seltene Erkrankungen sind zwei Kinderärzte - einer davon aus Münster: Der Pädiater Prof. Heymut Omran und sein…

Münster (mfm/mk) – Für Patienten, die an chronischen Entzündungen der Lungen und Nasennebenhöhlen leiden, gibt es neue Hoffnung. Ein Forschungsteam um den Direktor der Klinik für Allgemeine…