
Cystic fibrosis
Cystic fibrosis (CF) is a hereditary disease in which the gene encoding a chloride (CI) ion channel, cystic fibrosis transmembrance conductor regulator, CFTR, is mutated. The disease affects many different organs, but the most severe effects are found in the lungs, where the Cl channel is located on the surface of respiratory epithelial cells and regulates water balance, among other things. The mutation leads to a viscous secretion that surrounds the cells and causes the cilia on the cells to be unable to transport inhaled bacteria out of the respiratory tract. Therefore, the disease is associated with chronic recurrent bacterial infections that destroy the lung tissue over many years and thus impair the quality of life. One of the most common pathogens that can be detected from respiratory samples is Staphylococcus aureus (S. aureus). Because patients present with other symptoms besides bacterial respiratory infections, they are treated at specialised centres, most of which are visited every 3 months.
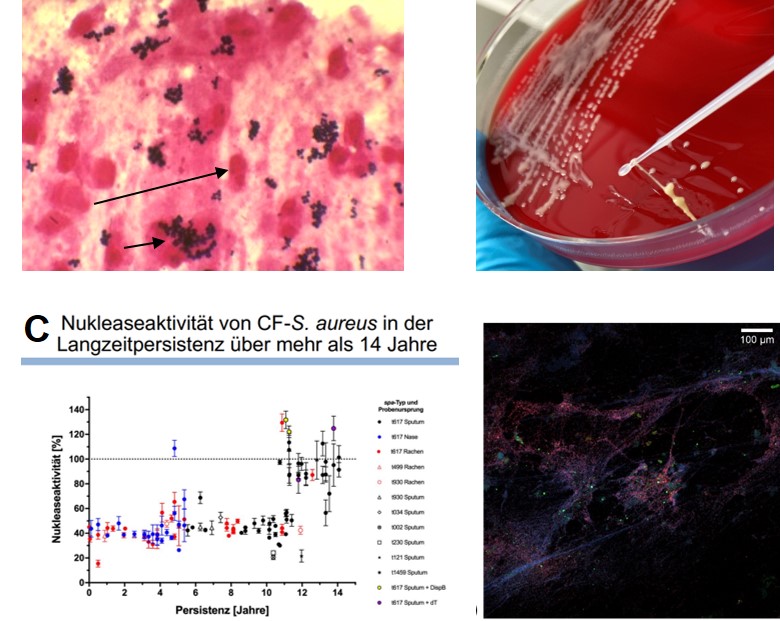
For more than 2 decades, we have been responsible for the microbiological analysis of samples from the respiratory tract and have since built up a biobank with several thousand S. aureus isolates from more than 200 patients (people with CF, pwCF). Using the sequential S. aureus isolates from individual pwCF, which we have analysed using different typing methods and functional experiments, we have been able to show that it is mostly the same S. aureus clone that has adapted over time to the hostile environment of the respiratory tract characterised by numerous leukocytes, other bacteria competing for food and repeated antibiotic therapies.