
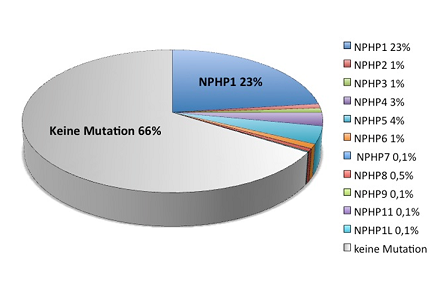
Für die Nephronophthise konnten bislang zumeist durch Positions-Klonierung Mutationen in 11 verschiedenen Genen identifiziert werden (NPHP1-11, NPHP1L) (siehe Tabelle2). Die häufigste Mutation stellt hierbei eine homozygote Deletion von NPHP1 dar, die in ca. 20% der Fälle mit isoliertem Nierenbefall nachgewiesen werden kann, während alle weiteren Genmutationen derzeit nur knapp 10% der Nephronophthise-Erkrankungen ausmachen. Bei etwa 70% aller Nephronophthise-Patienten ist die genetische Ursache noch ungeklärt.
Tab.2: Zusammenstellung der NPHP1-NPHP11-Gene, Genorte, renale und extrarenale Manifestationen, Häufigkeit der Mutationen.
Gen | Genort | Alter bei terminaler Nieren-insuffizienz , Median (Range) | Extrarenale Symptome | Mutations-Frequenz |
NPHP1 | 2q13 | NPH, 13 J (7-29 J) | RP (10%) | 23,4 % homozygote Deletion |
NPHP2/INVS
| 9q31 | NPH, (1-5 J) | RP (10%) | 1,4 % |
NPHP3
| 3q22 | NPH, 19 J (11-47 J) | LF | 0,7 % |
NPHP4
| 1p36 | NPH, 21 J (6-35 J) | RP (10%) | 2,6 % |
NPHP5/IQCB1
| 3q21 | NPH, 15 J (15J (6-32 J) | Early-onset RP | 3,6 % |
NPHP6/CEP290
| 12q21 | NPH, 12 J (5-17 J) | JS | 1 % |
NPHP7/GLIS2
| 16p | NPH | - | 0,1 % |
NPHP8/RPGRIP1L
| 16q | NPH | JS | 0,5 % |
NPHP9/NEK8 | 17q11 | NPH, (<5 J) | - | 0,1 % |
TMEM67/MKS3/ | 8q22.1 | NPH | JS | ? |
NPHP1L/XPNPEP3
| 22q13 | NPH | Kardiomyo-pathie | 0,1 % |
JS: Joubert-Syndrom, LF: Leberfibrose, MKS: Meckel-Gruber-Syndrom, OMA: okulomotorische Apraxie, RP: Retinitis pigmentosa, VSD: ventrikulärer Septum-Defekt