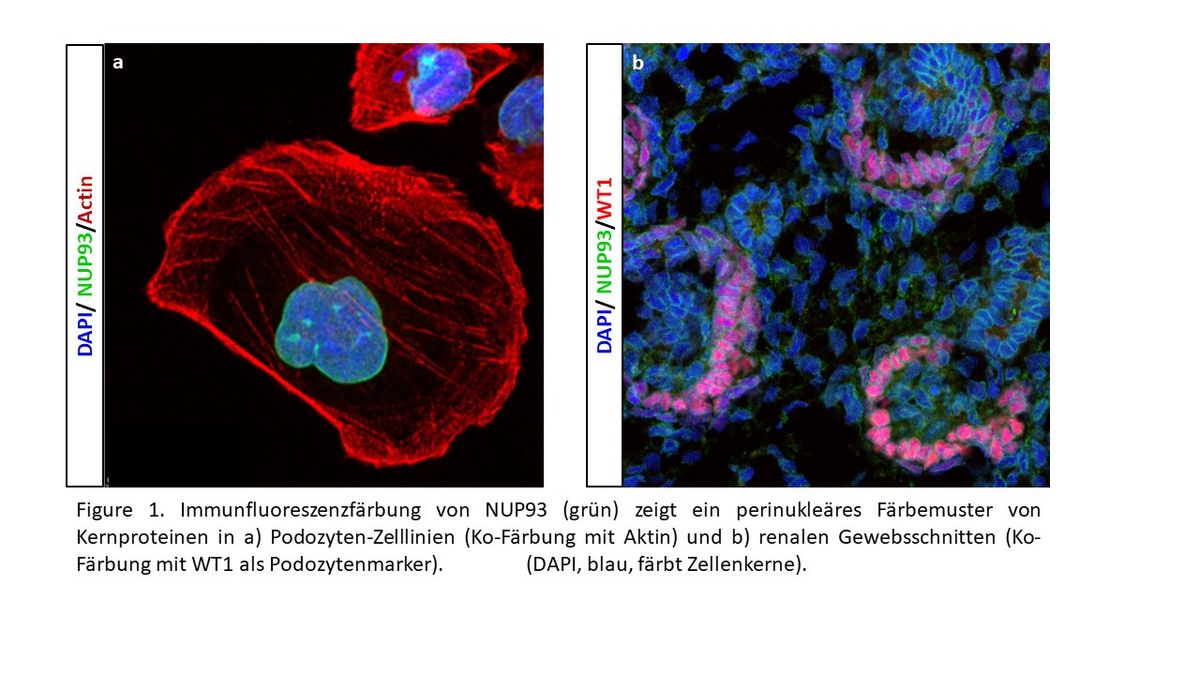
Point mutations in nuclear pore proteins, such as the nucleoporin NUP93, as a cause of inherited nephrotic syndrome
In genetic studies, we identified mutations in various structural proteins of the nuclear pore complex (NUP85, NUP93, NUP107, NUP133, NUP160, and NUP205), which cause early-onset and progressively worsening kidney disease in affected children. This disease is characterized by podocyte injury, nephrotic syndrome, and chronic kidney failure.
The nuclear pore is a selective transport channel between the nucleus and the cytoplasm and represents the only connection between these compartments in eukaryotic cells. The spatial separation of transcription and translation—two consecutive steps in the conversion of genetic information into proteins—is essential for enabling cells to respond to external stimuli and adapt to changing environmental conditions.
This essential and universal function of nuclear pores in all nucleated cells contrasts with our genetic findings, which reveal a kidney-specific phenotype in affected patients. We therefore hypothesize that certain NUP proteins may have an additional, cell type–specific function in podocytes that is independent of their canonical role within the nuclear pore. Alternatively, it is conceivable that podocytes, due to their post-mitotic nature, are particularly vulnerable to disturbances in nuclear pore function.
In our project, we aim to resolve this apparent contradiction and gain a better understanding of the molecular disease mechanism underlying nuclear pore–associated podocyte injury. To this end, we have developed cell culture systems that allow for inducible knockout of NUP proteins as well as live cell imaging to visualize nuclear transport in real time. Additionally, we are studying mouse models that replicate the human phenotype and are therefore well suited for translational research.