
Übersichtstabelle publizierter PCD-Gene
PCD ist eine genetisch heterogene Erkrankung, welche in der Regel autosomal rezessiv vererbt wird. Für autosomal rezessiv vererbte PCD-Varianten wurden >50 Gene entdeckt. Nur PIH1D3 ist auf dem X-Chromosom lokalisiert, weshalb Mutationen in diesem Gen bei Männern auch hemizygot krankheitsursächlich sein können.
Bestandteil des äußeren Dyneinarmes
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| DNAI1 | ODA-Defekt | Ja |
| DNAI2 | ODA-Defekt | Ja |
| DNAH5 | ODA-Defekt | Ja |
| DNAH9 | ODA-Defekt | Ja |
| DNAH11 | nicht nachweisbar | Ja |
| CCDC103 | ODA-Defekt | Ja |
| DNAL1 | ODA-Defekt | Ja |
| TXNDC3 (NME8) | partieller ODA-Defekt | Ja |
Bestandteil des ODA-Docking-Komplexes
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| CCDC114 | ODA-Defekt | Ja |
| ARMC4 | ODA-Defekt | Ja |
| CCDC151 | ODA-Defekt | Ja |
| TTC25 | ODA-Defekt | Ja |
Assemblierung der ODA- und IDA-Komplexe
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| LRRC50 (DNAAF1) | ODA- und IDA-Defekt | Ja |
| KTU (DNAAF2) | Partieller ODA- und IDA-Defekt | Ja |
| DNAAF3 | ODA- und IDA-Defekt | Ja |
| DYX1C1 (DNAAF4) | ODA- und IDA-Defekt | Ja |
| LRRC6 | ODA- und IDA-Defekt | Ja |
| HEATR2 | ODA- und partieller IDA-Defekt | Ja |
| ZMYND10 | ODA- und IDA-Defekt | Ja |
| SPAG1 | ODA- und IDA-Defekt | Ja |
| CFAP298 (C21orf59) | ODA- und IDA-Defekt | Ja |
| PIH1D3 | ODA- und partieller IDA-Defekt | Ja |
| CFAP300 (C11orf70) | ODA- und IDA-Defekt | Ja |
Bestandteil des Dynein-Regulator-Komplexes (DRC)
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| CCDC39 | Mislokalisation des zentralen Tubuluspaares; defizientes Assembly des IDA und des DRCs. | Ja |
| CCDC40 | Mislokalisation des zentralen Tubuluspaares; defizientes Assembly des IDA und des DRCs. | Ja |
| DRC1 (CCDC164 ) | Verlust der Nexin-Brücken (schwer detektierbar) | nicht belegt |
| DRC4 (GAS8) | Defizientes Assembly des DRC | nicht belegt |
| DRC2 (CCDC65) | Reduktion des IDA-Komplexes und der Nexin-Brücken |
Bestandteil des zentralen Proteinkomplexes
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| HYDIN | Verlust der "C2b"-Struktur (schwer detektierbar) | Nein |
| STK36 | normale Ultrastruktur | Nein |
| SPEF2 | normale Ultrastruktur | Nein |
Bestandteil der Radialspeichen
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| RSPH1 | Gestörte Lokalisation des zentralen Tubuluspaares | Nein |
| RSPH3 | Gestörte Lokalisation des zentralen Tubuluspaares | Nein |
| RSPH4A | Gestörte Lokalisation des zentralen Tubuluspaares | Nein |
| RSPH9 | Gestörte Lokalisation des zentralen Tubuluspaares | Nein |
| DNAJB13/RSPH16A | Gestörte Lokalisation des zentralen Tubuluspaares | Nein |
Weitere PCD-assoziierte Gene
| Name | Phänotyp (Ultrastruktur) | Situs inversus (~50%) |
| GAS2L2 | Störung der ciliären Orientierung | ? |
| LRRC56 | Abwesenheit des ODA im distalen Bereich des Axonem | Ja |
Einen X-chromosomal gekoppelten Vererbungsgang zeigen zwei weitere Gene: RPGR und OFD1. Mutationen in diesen Genen sind neben PCD auch mit anderen gravierenden Erkrankungen assoziiert.
Unsere Arbeitsgruppe war an der Identifizierung der mit einem Link versehenen Gene maßgeblich beteiligt. Wir forschen kontinuierlich nach neuen, für PCD ursächlichen Genen.
Vom Phänotyp zur Kandidatengenanalyse
Der Primären Ciliären Dyskinesie liegen Fehler der strukturellen und funktionellen Bestandteile der Cilie und assoziierter Proteine zugrunde. An dem koordinierten Cilienschlag des Flimmerepithels sind ca. 250 Proteine beteiligt. Mutationen in jedem korrespondierenden Gen dieser 250 Proteine könnten eine PCD bewirken. Die Gene sind teilweise sehr groß, die schwere Dyneinkette DNAH5 des äußeren Dyneinarmes (ODA) besitzt allein 80 kodierende Genabschnitte (Exone). Eine genetische Analyse ist deshalb sehr aufwendig, zumal noch nicht alle humanen Gene identifiziert sind.
Um neue Kandidatengene zu finden sind einige Informationen notwendig. Die Kopplungsanalyse (Vergleich der Allele von gesunden und erkrankten Familienmitgliedern) gibt Auskunft über den Genort, der Phänotyp über die mögliche Funktion des fehlerhaften Proteins. Zeigt sich in der Hochfrequenzanalyse z. B. ein steifer, zittriger Schlag weist dies auf einen Defekt des inneren Dyneinarmes hin; ein nur noch gelegentliches Zucken oder völlige Unbeweglichkeit deutet dagegen auf einen Defekt des ODA. Die Immunfluoreszenzanalyse (IF) ermöglicht genauere Hinweise auf die Funktion und Lokalisation des gesuchten Genproduktes. Ist ein Kandidatengen gefunden, weist die PCR-Amplifikation aller kodierenden Genabschnitte und bidirektionaler Sequenzierung die ursächliche Mutation nach.
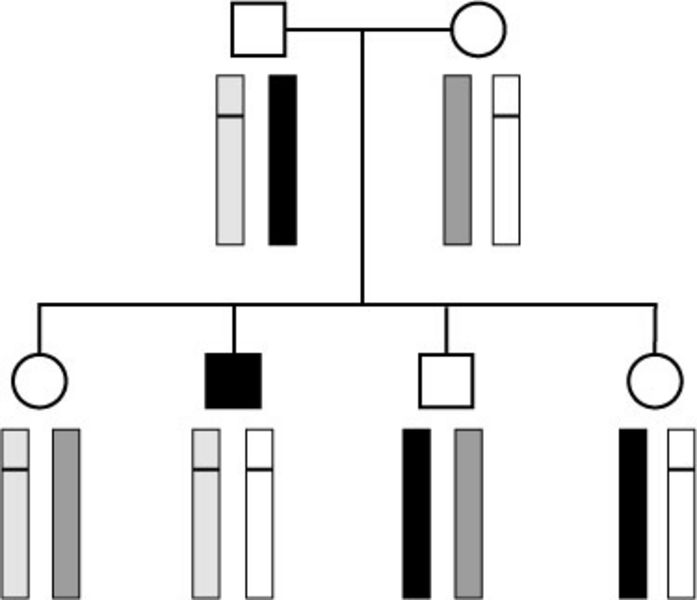
Autosomal rezessiver Erbgang: beide Elternteile haben zwei Kopien (Allele) von jedem Gen. Ist nur ein Gen durch eine Mutation verändert (mit einem Querstrich markiert) sind sie selber gesund, geben dieses Allel aber an ihre Kinder weiter. Die Kinder erhalten ein Allel von jedem Elternteil, bei zwei mutierten Allelen erkrankt das Kind.
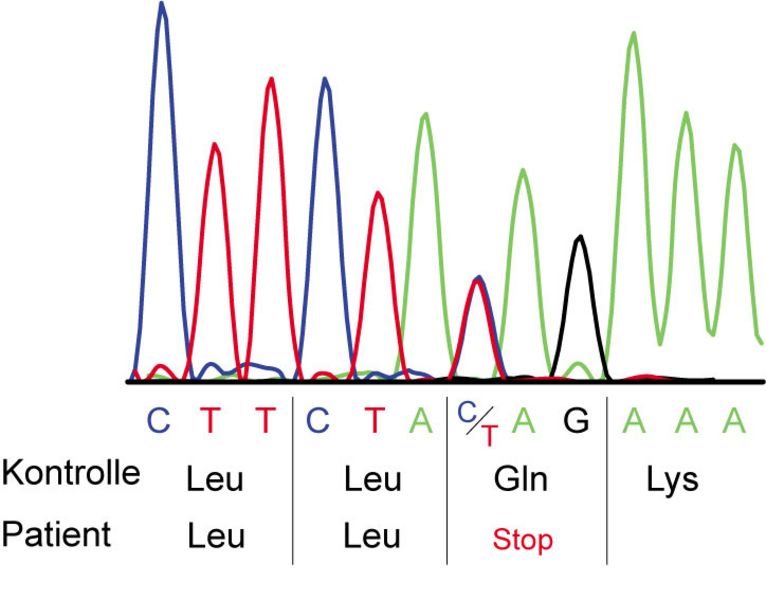
Sequenzanalyse des DNAH5-Gens, Abbildung einer heterozygoten STOP-Mutation. An einer Stelle des Transkriptes weist der Patient auf einem Allel eine Mutation auf, die zu einem Abbruch der Proteinherstellung führt. Die zweite Mutation vom anderen Elternteil liegt an einer anderen Stelle des Gens auf dem zweiten Allel.
Molekulargenetische Diagnostik
Wir führen molekulargenetische Diagnostik bekannter Gene bei Patienten mit PCD durch. Die Diagnose "PCD" wird von unserem Labor durch Immunfluoreszenzmikroskopie verifziert. Anhand der IF-Daten werden die zu untersuchenden Gene ausgesucht, z.B. werden die für Proteine des äußeren Dyneinarmes kodierenden Gene nur sequenziert, wenn auch ein ODA-Defekt vorliegt. Bitte denken Sie an die Ausstellung eines Laborüberweisungsscheines.
Die Teilnahme an unserer IF-Diagnostik ist deshalb Vorausetzung für die Durchführung der molekulargenetischen Analyse. Die Einverständniserklärung des Patienten beinhaltet auch die Teilnahme an unserer fortlaufenden Forschung. Weitere Informationen und Unterlagen finden Sie in den folgenden links oder wenden Sie sich direkt an unser Labor in Münster, wir senden Ihnen ein Kit zu.
Informationen und Unterlagen zur genetischen Analyse (Blutentnahme)
- Patienteninformation für Kinder und Jugendliche
- Information und Einwilligungserklärung Patient
- Information und Einwilligungserklärung für Eltern
- Einwilligung zur molekulargenetischen Diagnostik
Begleitbögen für die Blutprobe
Link zur IF-Diagnostik
Weiterführende Literatur