For the 3rd time, miGenomeSurv members met at the Bonifatiuskloster in Hünfeld to discuss new software features, experiences from the past year, recent scientific advances and future developments. As last year, they were accompanied by members of the WIN-KID consortium who complemented the program with perspectives from AI-based AMR prediction. We are looking forward to coming back in June 2027!
Network expands to whole "DACH"-region
We are happy to announce that the network was recently joined by the University of Zürich and thus now covers all three countries of the DACH region - Germany, Austria and Switzerland. This will further enhance our cross-border surveillance, leading to an earlier detection of international outbreaks.
Network meeting 2025
In good tradition, members of the network came together for a yearly exchange in May 2025. Again, we met at the Bonifatiuskloster in Hünfeld for two days of sharing experiences, discussing future developments, and networking. For the scientific input, this year we were joined by member of the WIN-KID consortium ( https://www.medizin.uni-muenster.de/win-kid/startseite.html ), who shared their insights about the use of machine learning techniques to predict antimicrobial resistance patterns. All active and interested member of the network are invited to join us for the next meeting in 2026!
Network meeting 2024
For the third time, members of the miGenomeSurv network came together for the yearly network meeting in June 2024. Located centrally within Germany, the Bonifatiuskloster Hünfeld, close to Fulda, offered a wellcoming atmosphere. Members of the network shared their experiences and new ideas. Moreover, the latest improvements in long-read sequencing technologies and analysis of mobile genetic elements were discussed. We are looking forward to coming back next year in May for the network meeting 2025.
Network Meeting 2023
After a successful kick-off meeting in 2022, the network participants reconvened in June 2023 for our yearly network meeting. This time, we met in Retz, a lovely town in Lower Austria with a huge labyrinth of historical wine cellars under its streets.
We welcomed our new participants, shared experiences, and discussed plans for the future. Our scientific program included studies on thresholds for cluster detection, surveillance of different species in Austria and Germany, the Swiss Pathogen Surveillance Platform, and recent advances in long-read sequencing technologies.
We really enjoyed the fruitful discussions within and between the sessions and look forward to meeting again in 2024!
Network partners published paper on multicountry listeriosis outbreak
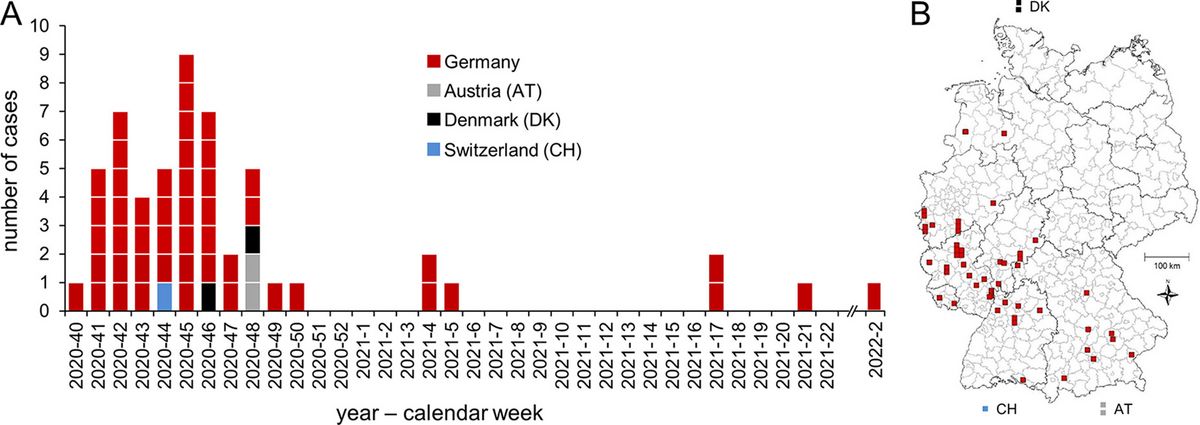
Chronology of outbreak progression and geographical distribution of cases.
Scientists from the miGenomeSurv network partners RKI and AGES have published a paper on a large multicountry outbreak of Listeria monocytogenes. The outbreak involved 55 cases in Germany, Austria, Denmark, and Switzerland in 2020-2021. It was linked to smoked rainbow trout filets. Outbreak isolates from both Germany and Austria were deposited in miGenomeSurv and led to matching alerts in both institutions. Read the full paper here: Halbedel et al., Microbiol. Spectr., 2023.
Next miGenomeSurv network meeting in June 2023
After our successful kick-off meeting in Northeim in June 2022, we are happy to announce that we will reconvene on the 15th/16th June 2023 for our second network meeting. Organized by the academy of the co-founders AGES, we will meet in Retz, a beautiful small town close to Vienna. We are looking forward to two days full of fruitful discussions and scientific exchange For questions regarding the meeting please contact us.
In June 2022, representatives of the founding organizations and network participants came together in Northeim to initiate the miGenomeSurv network in a kick-off meeting.
Accompanied by invited speakers, the group spent 2 fruitful days with scientific talks, experience reports, and discussion sessions. During a BBQ evening in the beautiful surroundings of Lower Saxony’s forests, the participants were able to get to know each other and build foundation for successful cooperation within the network.
We are looking forward to our next meeting in June 2023. Further information will follow soon!
(Picture taken before the Corona pandemic.)
Datum: 02.06.2021 (German version below)
Network uses "genetic fingerprint" to monitor bacterial pathogens and to expedite the detection of outbreaks.
In a modern and global society, the mobility of humans and animals, and the transport of goods and food are on a permanent high. Infectious diseases and their causative agents have always followed trade and travel routes and the contact points involved. This is impressively illustrated by the current SARS-CoV2 pandemic. Further in 2011, the German public health system was confronted with a food-borne outbreak of a pathogen - an E. coli EHEC O104: H4 variant - which caused one of the largest outbreaks of EHEC infections resulting in 2,987 cases of diarrhea, 855 cases of severe kidney failure and 53 deaths.
The identification of the source of infection is usually essential for successful outbreak control. If the infection source is not identified quickly, such outbreaks can go on for long periods of time and across different locations, making the identification of the causative agent very difficult.
However - how do you recognize that the pathogens involved are "similar"? What does "similar” mean, and how can we identify whether a pathogen has already been observed elsewhere or earlier? How exactly can pathogens be traced? For this, a detailed "fingerprint" of the pathogen is needed which identifies or excludes any similarities.
"In order to improve infection control, molecular surveillance of infectious agents is essential", says Professor L. H. Wieler, President and Head of the Robert Koch Institute. To provide suitable instruments a consortium has been initiated by partners from three German organizations: the University of Münster, the Research Center Borstel and the Robert Koch Institute to form the network "miGenomeSurv" (microbial genome-based surveillance of infectious agents). This network is based on national reference laboratories, where - according to their mandate - infectious agents relevant to the population are characterized not just microbiologically but also via genome analysis. Genome sequencing methods provide "fingerprints" and other characteristics of the bacteria allowing surveillance and cluster detection.
In the context of "miGenomeSurv", a common language (nomenclature) is defined for the numerous lineages of detected pathogens. "Easy-to-use bioinformatic tools help to give each pathogen a unique signature", explains Professor Dag Harmsen of the University of Münster. This particularly important aspect is based on the genome sequence and of a so-called core genome (cg) –MLST type calculated from the genome data. As a result, the genetic material of the pathogen is translated into a standardized numerical code. This allows an easy exchange of data between the participating laboratories, with other national and international partners and institutions such as the ECDC and the public health service responsible for implementing measures such as outbreak management. The genomic profiles and other project data are published in a summarized form on the network’s website at www.miGenomeSurv.org.
The consortium will first concentrate on the following high-priority organisms:
enterohaemorrhagic Escherichia coli (EHEC)
Listeria monocytogenes
multi-resistant Mycobacterium tuberculosis, M. bovis / caprae
Vancomycin-resistant enterococci (VRE).
Ausbrüche schneller erkennen - Netzwerk nutzt „genetischen Fingerabdruck“ für die Überwachung (Surveillance) von bakteriellen Krankheitserregern
In der modernen, global agierenden Gesellschaft ist die Mobilität von Menschen, Tieren, Waren und Lebensmitteln extrem hoch. Stets folgten Infektionen und deren Erreger den Handels- und Reisewegen und dem damit verbundenen Kontakt. Dies verdeutlicht insbesondere die gegenwärtige SARS-CoV2 Pandemie. Ein weiteres prägnantes Beispiel findet sich im Jahr 2011. Hier wurde das deutsche Gesundheitssystem mit dem lebensmittelbedingten Ausbruch eines Krankheitserregers - einer toxischen Variante des Darmbakteriums E. coli des Typs EHEC O104:H4 - konfrontiert, welcher zum weltweit größten EHEC-Ausbruch mit 2.987 Fällen von Durchfall, 855 Fällen von schwerem Nierenversagen und 53 Todesfällen führte.
Die Identifizierung der Infektionsquelle ist in der Regel Voraussetzung für eine erfolgreiche Ausbruchsbekämpfung. Wird die Quelle nicht zeitnah identifiziert, können solche Ausbrüche auch über lange Zeiträume und an verschiedenen Orten auftreten, was die Identifizierung eines Zusammenhangs besonders schwierig macht.
Aber - wie erkennt man, dass die Erreger „gleichartig“ sind? Was heißt „gleichartig“, und wie lässt sich erkennen, ob ein Erreger bereits an anderem Ort und zu anderer Zeit einmal beobachtet wurde? Wie lässt sich die Spur von Krankheitserregern exakt verfolgen? Hierfür wird ein exakter „Fingerabdruck“ des Erregers benötigt, um Ähnlichkeiten und Verwandtschaften zu erkennen oder auszuschließen.
„Um den Infektionsschutz zu verbessern, ist eine molekulare Surveillance von Infektionserregern unverzichtbar“, sagt Professor L. H. Wieler, Präsident des Robert Koch-Institutes. Um hierfür geeignete Instrumente bereitzustellen hat sich eine Gruppe von Wissenschaftlern an der Universität Münster, dem Forschungszentrum Borstel und dem Robert Koch-Institut im Netzwerk „miGenomeSurv“ - für mikrobielle genombasierte Surveillance von Infektionserregern - zusammengeschlossen. Basis des Netzwerks sind die dort angesiedelten nationalen Referenzlabore, bei denen - ihrem Auftrag entsprechend - für die Bevölkerung relevante Infektionserreger mikrobiologisch und bis hin zur Erbgutanalyse mittels Genomsequenzierung charakterisiert werden, um hieraus den „Fingerabdruck“ und weitere Merkmale der Bakterien für die Erregerüberwachung und Ausbruchsaufklärung zu gewinnen.
Im Rahmen von „miGenomeSurv“ wird eine einheitliche „Sprache“ für die zahlreichen Erregerlinien und die Erstellung eines „Steckbriefes“ genutzt. „Einfach zu verwendende bioinformatische Werkzeuge helfen dabei, dem jeweiligen Erreger eine eindeutige Signatur zu geben“, erläutert Professor Dag Harmsen von der Universität Münster. Dieser besonders wichtige Aspekt einer abgestimmten „Sprachregelung“ beruht auf der Berechnung eines sogenannten Kerngenom (Core Genome-(cg))-MLST-Ansatzes/Schlüssels aus den Genomdaten. Hierdurch wird das Erbgut der Erreger in einen standardisierten Zahlencode übersetzt. Das ermöglicht einen reibungslosen Datenaustausch zwischen den beteiligten Laboratorien, mit weiteren nationalen und internationalen Partnern und Institutionen wie z.B. dem ECDC und dem für die Umsetzung von Maßnahmen zuständigen öffentlichen Gesundheitsdienst. Die genomischen Profile sowie weitere Daten des Projekts werden in zusammengefasster Form auf der Webseite des Netzwerks unter www.miGenomeSurv.org veröffentlicht.
Die Kooperationspartner konzentrieren sich zunächst auf die folgenden Erreger und stellen die dazugehörigen Daten zur Verfügung:
Enterohämorrhagische Escherichia coli (EHEC)
Listeria monocytogenes
Multidrug resistant Mycobacterium tuberculosis, M. bovis/caprae