
Since the 1980s, a few case reports described patients with respiratory “ciliary aplasia” or “acilia syndrome.” We analyzed “ciliary aplasia” candidates (including patients from previous reports) and identified recessive mutations in CCNO (encoding Cyclin O) and MCIDAS (encoding Multicilin) in a large cohort of individuals, respectively. All individuals suffered from severe respiratory symptoms of the upper and lower airways and development of bronchiectasis at an early age. Analysis of respiratory epithelial cells obtained by nasal brush biopsy by both transmission electron microscopy (TEM) and immunofluorescence analysis (IF) revealed that respiratory cilia were not completely absent; some cells still retained one or two cilia. These cells not only showed a low number of cilia documented by TEM and IF, but also a reduction and mislocalization of basal bodies and rootlets throughout the cytoplasm. Detailed analyses in both man and Xenopus revealed that this reduction of cilia number was due to a centriole amplification defect in the acentriolar pathway, which is specific for multiciliated cells. Further analyses demonstrated that MCIDAS functions upstream of CCNO and FOXJ1, which is important for transcriptional control of axonemal motor proteins such as DNAH5 and CCDC39. Whereas cilia in CCNO-mutant cells still contain motility-related proteins and can display normal beating patterns, MCIDAS-mutant cells are immotile and lack those axonemal motor proteins. MCIDAS and CCNO lie adjacently on chromosome 5q11 in a region related to multiciliogenesis, and act in the same pathway underlying multiciliogenesis. Based on these findings, we propose that this disease now should be referred to as “mucociliary clearance disorder with reduced generation of multiple motile cilia” (RGMC).
FOXJ1 belongs to the group of forkhead transcription factors, which are important for the ciliogenesis of motile cilia. Heterozygote de novo mutations cause a reduction of motile cilia and thus a ciliopathy characterized by internal hydrocephalus, chronic severe respiratory diseases and randomization of left and right body asymmetry.
FOXJ1 mutations lead to a clinical picture belonging to the group of primary ciliary dyskinesia. The autosomal dominant inheritance and the associated hydrocephalus are extraordinary.
| Name | Phenotype (ultrastructure) | Situs inversus (~50%) |
| CCNO | reduction of cilia | No |
| MCIDAS | reduction of cilia | No |
| FOXJ1 | reduction of cilia, hydrocephalus | Yes |

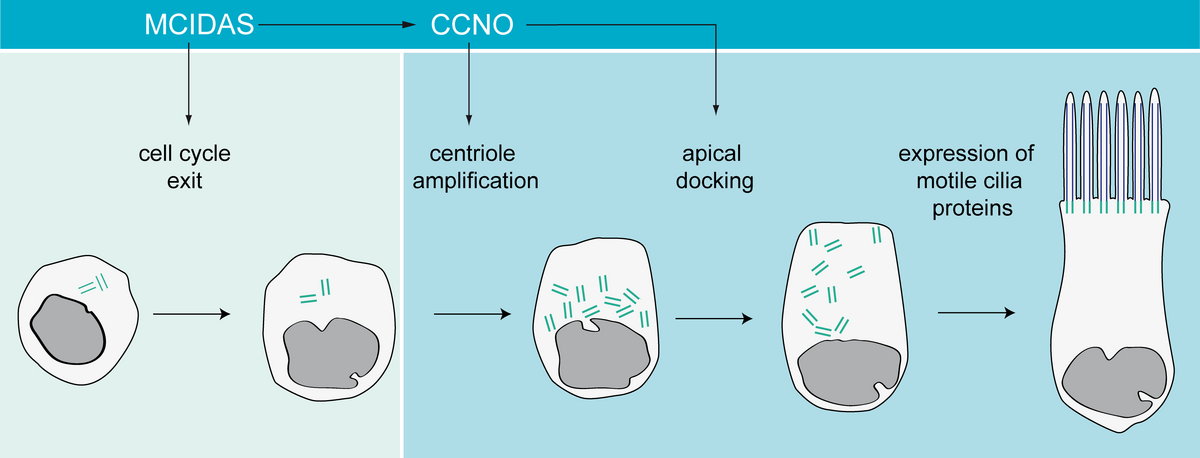
Differentiation of a multiciliated respiratory cell.