News
JAM-A and cell cannibalism
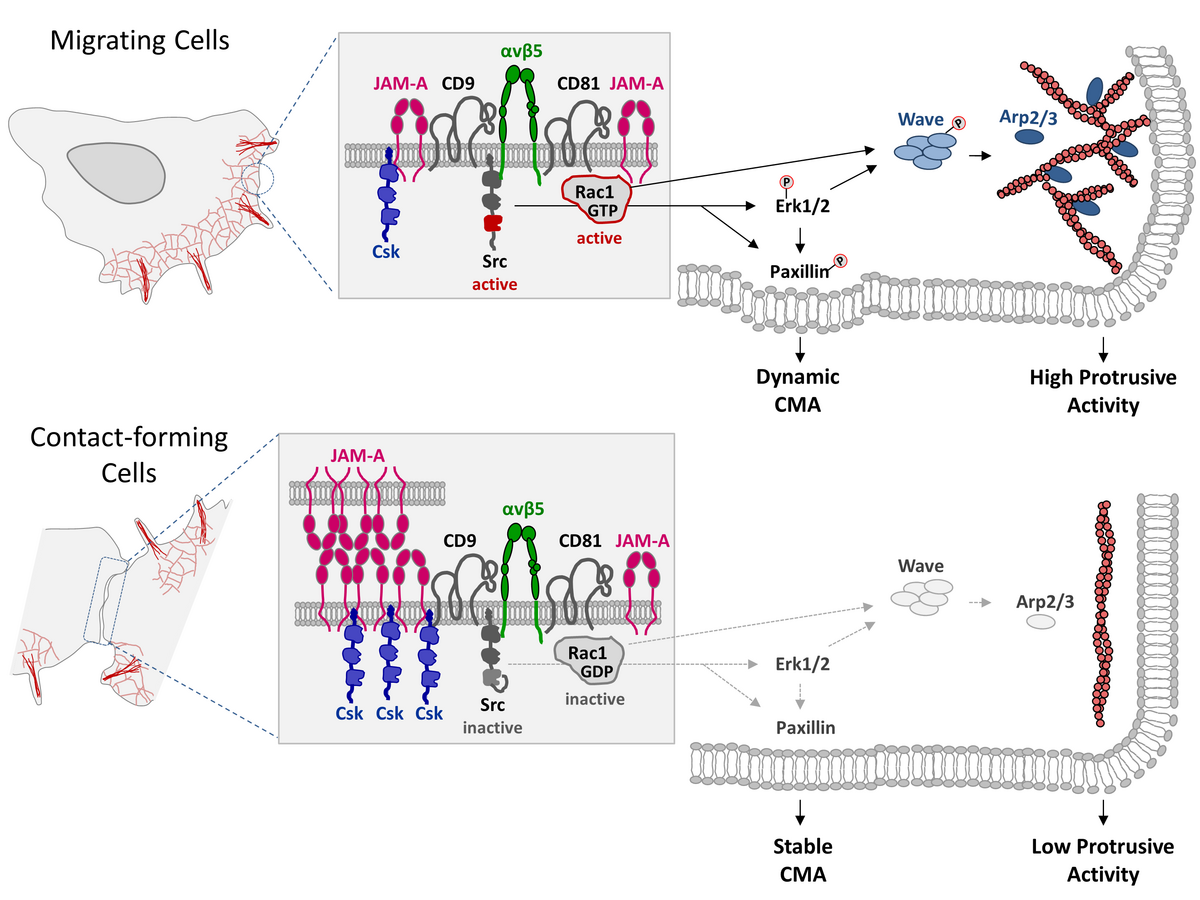
In our recent study we describe in more detail an observation that we made during an earlier study. In that study we had found that JAM-A limits cell motility and triggers a process called “contact inhibition of locomotion” (CIL) when migrating interact with other cells. This process is required to prevent cellular overgrowth and to ensure monolayer formation (see “News” section below: “JAM-A makes contact inhibition”).
When we analyzed cells migrating on micropatterns we observed that JAM-A-depleted cells not only migrated across other cells but formed cell-in-cell structures, as if cells would feed on each other through cell cannibalism. Cell-in-cell structures between non-phagocytic cells are frequently observed, and various types of cell-in-cell structures are described in the literature.
We characterized the type of cell cannibalism observed after JAM-A depletion as entosis. During entosis, tumor cells internalize other tumor cells to get access to the energy resources of the internalized cell. The onset of entosis after JAM-A depletion strongly correlated with a loss of CIL and involves the same molecular mechanism.
We found that the suppression of entosis by JAM-A correlates with the ability of JAM-A to regulate CIL, and that it involves the same molecular mechanisms. Our observation, thus, indicate that JAM-A not only regulates inhibition of cell motility in response to cell-cell contact formation but that a loss of CIL is linked to a process of cell cannibalism called entosis. This study is published in iScience.
Two MCF7 tumor cells – one JAM-A KD cell (green) and one wildtype cell (red) – in a fierce battle. Note that the red cell repeatedly escapes from the green cell’s clinch. In the majority of cases, the JAM-A KD cells ended up as the winner and degraded the wildtype cell within a vacuole.
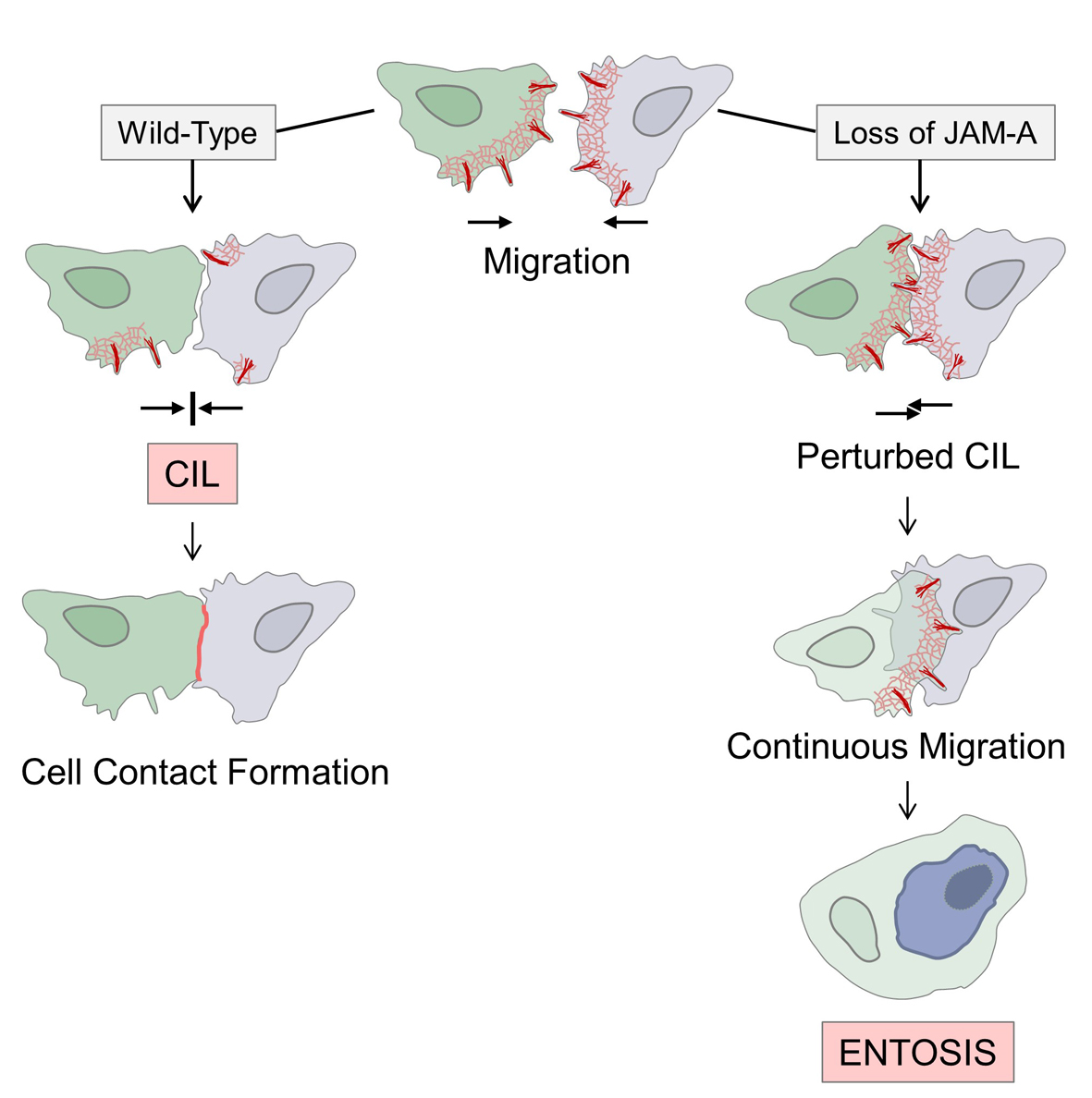
Left (wildtype condition): Contact-forming migrating cells downregulate protrusive activities (normal CIL response) and form cell-cell contacts. Right (JAM-A knockdown condition): Contact-forming cells fail to downregulate protrusive activities (no CIL response) and continue migration. In many cases, one of the two cells cell is engulfed by the other cell through entosis.