
News
Web-App hilft beim Erbgutabgleich von Säugetieren
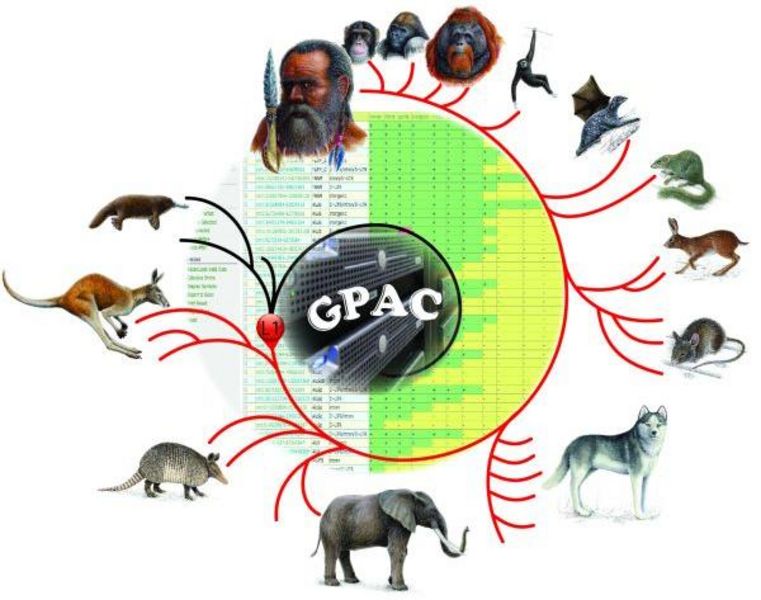
Mit dem GPAC-Tool lassen sich genomische Sequenzeinschübe zwischen unterschiedlichen taxonomischen Gruppen analysieren (hier eine von dem Forscherteam erstellte und als MBE-Titelbild ausgewählte schematische Darstellung)
Münster (mfm/tw) – Von Mäusen und Menschen: Auch Säugetiere, die ganz unterschiedlich aussehen, können viel gemein haben. Um die Verwandtschaft zu klären, hilft ein Blick ins Erbgut. Eine neue Webanwendung aus der Universität Münster ziert nun den Januar-Titel der Fachzeitschrift „Molecular Biology and Evolution“ – weil sie hilft, tausende Erbgutsequenzen auf einmal abzugleichen.
Wie Tiere miteinander verwandt sind, ist oft umstritten. Das Auge führt leicht in die Irre: Spitzmäuse etwa sehen den Mäusen zwar ähnlich, stehen dem Igel aber näher. Die Bioinformatik hilft, solche Fragen zu klären. Sie kann auf riesige Bibliotheken mit den vollständigen Erbanlagen vieler Säugetiere zurückgreifen, die in den letzten eineinhalb Jahrzehnten entschlüsselt worden sind; auch das Genom des Menschen ist seit 2001 genau bekannt.
Um Verwandtschaften zu klären, die Evolution neuer Gene zu ergründen oder krankheitsauslösende Einschübe oder Sequenzverluste zu ihrem Ursprung zu verfolgen, haben Wissenschaftler um den Privatdozenten Dr. Jürgen Schmitz und seine Doktorandin Angela Noll vom Institut für Experimentelle Pathologie der Medizinischen Fakultät der Universität Münster den „Genome Presence/Absence Compiler“ (GPAC) entwickelt. Die Web-App entstand als Teil von Nolls Doktorarbeit in Zusammenarbeit mit Norbert Grundmann vom Institut für Bioinformatik. „Wir nehmen eine Sequenz aus dem menschlichen Erbgut und schauen, ob sie bereits im Schimpansen, der Maus, dem Hund oder gar im Schnabeltier vorhanden ist“, erläutert Schmitz das Prinzip: „Wenn wir sie auch bei engen Verwandten nicht finden, haben wir sie vermutlich im Laufe unserer individuellen Evolution erworben. Ansonsten können wir abschätzen, zu welchem Zeitpunkt der Evolutionsgeschichte sie zum ersten Mal aufgetreten ist.“
Ein Tool, das diese Aufgabe erfüllt, gibt es bereits von der amerikanischen University of California, Santa Cruz (UCSC). Allerdings kann damit immer nur jeweils ein bestimmter Teil der Erbgutsequenz abgeglichen werden, die münstersche App verarbeitet tausende Abfragen auf einmal; in einer Tabelle wird übersichtlich ausgegeben, welche Sequenz sich bei welchen Tieren findet. Und obwohl nur das Erbgut rezenter Arten verglichen wird, führen die Ergebnisse manchmal weit in die Vergangenheit, so Schmitz: „Wenn die gleiche Sequenz bei sehr weitläufig verwandten Säugetieren auftritt, nicht jedoch in Reptilien, hatte sie vermutlich schon der letzte gemeinsame Säuger-Vorfahre – ein Urahn, der vor mehr als 160 Millionen Jahren gelebt hat.“ So kann mit wenigen Mausklicken getestet werden, wann neue Eigenschaften oder krankheitsauslösende Veränderungen in unserem Genom aufgetaucht sind und welche Veränderungen sie im Vergleich zu anderen Tierarten mit sich gebracht haben.
Publikation
Das Tool steht online bereit unter der URL: https://retrogenomics.uni-muenster.de/tools/gpac/index.hbi?lang=en
This could be interesting for you too:

Münster (mfm/mk) – Die Lebererkrankung Hepatitis B ist mit 350 Millionen chronischen Erkrankungen eine der häufigsten Virusinfektionen weltweit - und die Ahnen des Auslösers existieren…

Münster (mfm/mk) – Die Heimat der Koboldmakis sind die südostasiatischen Inseln, aber auch Zoobesucher kennen die „putzigen“ Baumbewohner mit den auffälligen großen Augen. Der Gang zu ihnen…

Münster (mfm) - Der diesjährige Bernhard-Rensch-Preis der Gesellschaft für Biologische Systematik (GfBS) geht an Alexander Suh, ehemaliger Doktorand der Westfälischen Wilhelms-Universität…

Münster (mfm/lwl) - Falken sind keine Greifvögel – jedenfalls stammesgeschichtlich, sondern sie sind enge Verwandte der Papageien und Sperlingsvögel. Und der gemeinsame Urahn von Papageien…
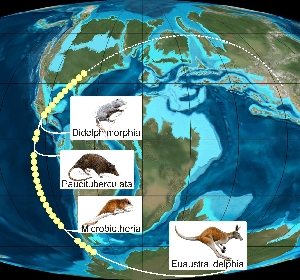
Münster (mfm/tw) – Ahnenforschung bei Skippy und seinen Verwandten: Ein Team um Dr. Jürgen Schmitz hat erstmals ein einfaches Modell für die Einwanderung der Beuteltiere nach Australien…